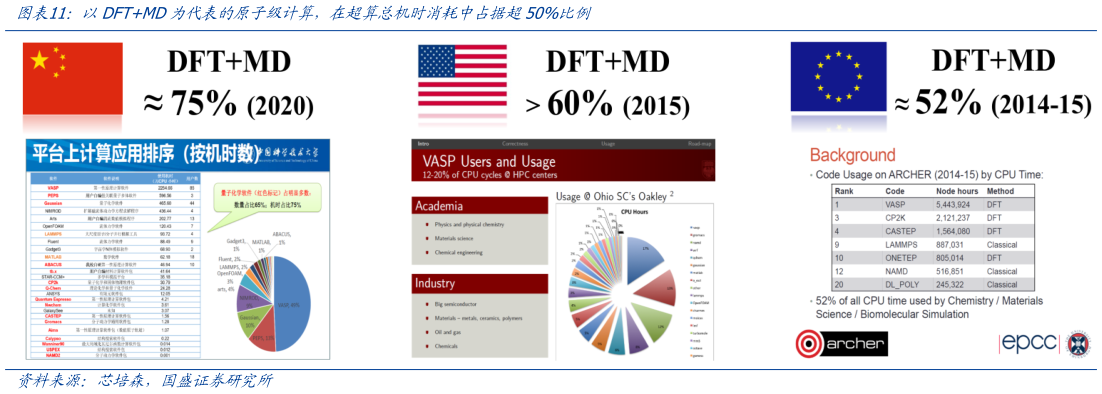
> 数据图表一起讨论下以DFTMD为代表的原子级计算,在超算总机时消耗中占据超50%比例2025-8-2以 DFTMD 为代表的原子级计算,在超算总机时消耗中占据超 50%比重。长久以来,分子动力学模拟因计算效率低下的问题,难以在业务场景中国用。举例来说,人体 典型的大分子蛋白质往往由几十万到上百万个原子构成,假设用分子动力学模拟方法计算模拟一个有着 50 万原子的蛋白质的 0.001 秒的瞬间动态,即使动用 10000 颗 CPU 并行计算,也需要耗费超过 100 年的时间。而人体 有超过 2 万种几十上百万个原子构成的人源蛋白质。由于 DFTMD 原子级计算要求极高,因此广泛采用超级计算机进行计算。根据芯培森,中科大在 的中、美、欧超算中心,以 VASP 为代表的量子化学计算 package占超算总机时消耗极高(中科大 75%,2020美国 Oakley 60%,2015欧洲 ARCHER 52%,2014-2015)。 VASP在多个超算中心均耗时第一。VASP是维也纳大学Hafner小组开发的进行电子结构计算和量子力学-分子动力学模拟软件包,是目前材料模拟和计算物质科学研究中流行的商用软件之一。VASP的基本原理是在密度泛函理论(DFT)框架 采用平面波基组展开的方法求解Kohn-Sham方程。VASP采用周期性边界条件(或超晶胞模型),基于密度泛函和赝势理论应原子、分子、表面、团簇等多种体系进行几何结构优化得到稳定构型,进而获得各种结构参数和能量,同时还能够计算多种电学、光学、磁学等性质。国盛证券科技传媒